Химическая энциклопедия
"РЕАКЦИОННАЯ СПОСОБНОСТЬ"
Главная - Химическая энциклопедия - буква Р - РЕАКЦИОННАЯ СПОСОБНОСТЬ
РЕАКЦИОННАЯ СПОСОБНОСТЬ
,
характеристика относительной хим. активности молекул, атомов, ионов, радикалов.
Для количеств. оценки Р.с. рассматривают реакционные серии, т.е. ряды однотипных
р-ций, проводимых в одинаковых условиях, напр.: (стандартная р-ция) где Х-общая группа атомов,
к-рая претерпевает изменения в данной р-ции (реакционный центр), Z0,
Zl,..., Zn-неизменяющиеся мол. фрагменты, Р0,
Р1,..., Рn -продукты р-ции. Отношения констант
скоростей k1/k0,..., kn/k0
количественно характеризуют Р. с. в ряду реагентов ZiX
(i = 0, 1, ..., п). В правильно составленной реакц. серии
изменение механизма реакции
должно быть исключено, т. е. константы скорости
должны характеризовать одну и ту же элементарную р-цию. Типичные реакционные
серии. Простейшая ситуация возникает при анализе изомерного состава продуктов
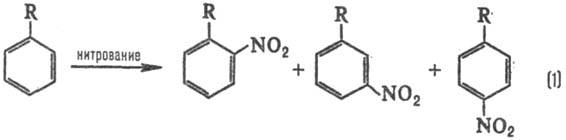
р-ции. В р-ции электроф. замещения в ароматич. ряду в зависимости от заместителя
R образуются те или иные изомеры, напр. при нитровании: Электронодонорные заместители
[R=СН3, ОСН3, N(CH3)2] стимулируют
образование орто-
и пара
-продук-тов, а электроноакцепторные (R
= СООН, SO3H, NO2)-мета
-продуктов, причем в первом
случае р-ция идет легче, чем с незамещенным бензолом (R = Н), а во втором-труднее.
Эти закономерности наз. правилами ориентации в ароматическом ряду. При нуклеоф.
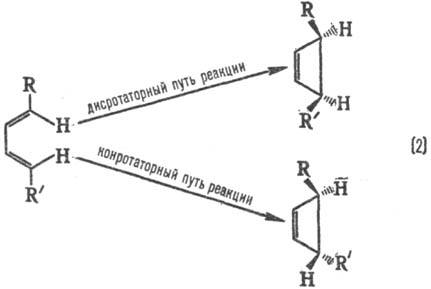
замещении правила ориентации обращаются. Стереохим. направленность
перипиклич. р-ций определяется Вудворда-Хофмаиа правилами, напр.: При дисротаторном пути
р-ции заместители R и R в продукте будут расположены по одну сторону плоскости
цикла, при конротаторном пути-по разные стороны. Эксперимент показывает, что
термич. циклизация производных бутадиена происходит по конротаторному пути,
а фотохим. циклизация-по дисротаторному пути. В примерах (1) и (2) нет
необходимости в количеств. кинетич. измерениях, Р. с. определяется по относит.
выходу изомеров. Пример широкой реакц. серии-р-ции радикального присоединения
по двойной связи: Р. с. характеризуется отношением
константы скорости k к константе скорости k0 р-ции
с этиленом (R, R = Н) (см. табл.). Аналогичные кинетич. измерения сделаны для
р-ций присоединения метильного радикала к ароматич. молекулам и для р-ций присоединения
др. радикалов. 
Квантовохимическая теория
Р.с. Совр. теоретич. химия позволяет непосредственно рассчитать абс. константы
скорости только для несложных хим. систем. В теории Р. с. качеств. закономерности
м.б. выявлены для объектов любой сложности. При этом используют разл. подходы.
При эмпирич. подходе классифицируют влияние заместителей по неск. типам (эффекты
сопряжения, полярные, пространственные и др.) и применяют корреляционные соотношения
. Традиц. квантовохим. подход основан на активированного комплекса теории
; при этом предполагается, что для всех р-ций, составляющих реакц.
серию (без пространств. и соль-ватац. эффектов), остается примерно постоянным
пред-экспоненц. множитель А в Аррениуса уравнении
для константы
скорости k = Aexp(-E
./RT) (R-газовая
постоянная, Т-абс. т-ра). Поэтому характеристикой Р.с. служит энергия активации
р-ции E., к-рая практически совпадает с высотой потенц.
барьера на поверхности потенциальной энергии
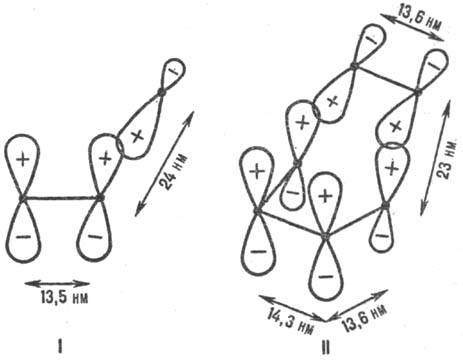
(ППЭ). Рис. 1. Переходные состояния
р-ций присоединения с участием p-электронных систем. I-радикальное присоединение
(метальный радикал + этилен), II-р-ция Дильса-Альдера (этилен + бутадиен); внизу
изображены соответствующие модельные p-сопряженные структуры. Показаны
атомные 2pp-орбитали, асимметрия к-рых обусловлена изменением
гибридизации вследствие взаимод. в переходном состоянии. Черные точки-ядра атомов
С. Знаки " + " и " —" относятся к соответствующим волновым
ф-циям. Орбитали перекрываются в фазе. Приведены межатомные расстояния в переходном
состоянии. Энергия стабилизации. Для
относит. оценки E. в реакц. сериях важна концепция делокализации
электронов в переходном состоянии (активир. комплексе) р-ции (М. Эванс, 1939).
Согласно этой концепции, электронное строение мн. реакц. центров подобно строению
сопряженных открытых и циклических я-электронных систем (рис. 1). Открытое переходное
состояние радикальной р-ции типа (3) подобно p-электронной системе аллильного
радикала (рис. 1, I), а "циклическое" переходное состояние согласованной
р-ции Дильса-Альдера (этилен + бутадиен) по строению напоминает бензол (рис.
1, И). p-Электроны заместителей R и R включаются в общую делокализованную
систему электронов. Относит. мерой энергии активации р-ции, а следовательно
и Р. с., может служить энергия стабилизации переходного состояния (энергия делокализации),
т.е. разность между энергией сопряженной системы p-электронов переходного
состояния и энергией p-электронов в изолир. реагентах. Энергию стабилизации
можно рассчитать полу-эмпирическими методами квантовой химии, напр. методом
Хюккеля. Вычисляя энергию стабилизации, можно предсказывать Р. с. хим. соединения
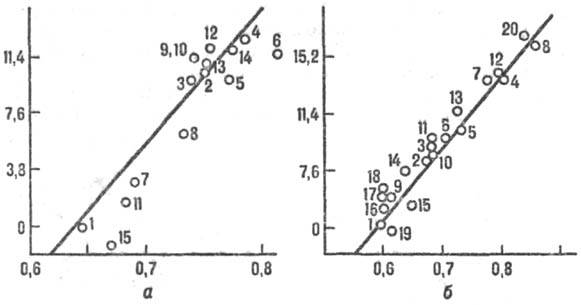
во мн. р-циях (иллюстрации см. на рис. 2). Рис. 2. Корреляция между
реакц. способностью и энергией стабилизации переходного состояния, а-р-ции
присоединения метального радикала СН•3 к олефи-нам, цифры
соответствуют номеру олефина в таблице. На оси ординат отложены величины 2,3
RТ lg k/k0 (кДж/моль), где k/k0 -
отношение констант скорости рассматриваемой р-ции и р-ции с этиленом, R-газовая
постоянная, Т-абс. т-ра. На оси абсцисс отложены энергии стабилизации в относит
единицах, б-р-ция СН•3 + ароматич. молекула. В
скобках указано положение в молекуле субстрата, для к-рого приводится константа
k. k0- константа скорости р-ции с бензолом. 1-бензол; 2-нафталин
(1); 3-фенантрен (9); 4-антрацен (9); 5-пирен (1); 6-хризен (6); 7-бенз(а)антрацен
(7); 8-нафтацен (5); 9-пиридин (2); 10-хино-лин (4); 11 -изохинолин (1); 12-акридин
(9); 13-феназин (1); 14-бензонитрил (4); 15-ацетофенон (4); 16-фторбензол (4);
17-хлорбензол (4); 18-бромбензол (4); 19-анизол (4); 20-бензохинон. Концепция граничных орбиталей.
Оценки Р.с. особенно просты, если использовать возмущений теорию
. В распространенном
варианте теории возмущений энергия стабилизации представляется в виде суммы
вкладов от взаимод. между мол. орбиталями реагентов. Наиб. вклад в сумму дают,
как правило, взаимод. граничных орбиталей, т.е. высших заполненных электронами
и низших незаполненных орбиталей; согласно К. Фукуи (1952), существенны только
эти вклады (см. Граничных орбиталей теория
). Концепцию граничных орбиталей
часто применяют в качестве основы для обсуждения Р. с. Альтернантные системы.
Качеств. подход, не обязательно использующий теорию возмущений, сформулирован
для класса сопряженных систем, наз. альтернант-ными. Они образованы из одинаковых
атомов (обычно углерода) и не содержат нечетных циклов (см. Альтернантные углеводороды
). Для таких систем в рамках Хюккеля
метода можно без всяких вычислений выявить нек-рые общие закономерности. Так, введение
полярного заместителя приводит к чередованию положит. и отрицат. изменений электронной
плотности в сопряженной углеродной цепи относительно незамещенного углеводородного
соед. (закон альтернирующей полярности, Ч. Коул-сон, Г. Лонге-Хиггинс, 1947).
Этот вывод теории позволяет объяснить правила ориентации в ароматич. ряду (р-ция
1). Для циклич. переходного
состояния (активир. комплекса) существенно, каким образом замыкаются новые связи:
в фазе или в противофазе, т.е. имеют ли атомные орбитали реагентов в области
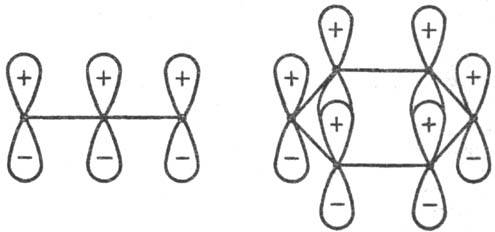
макс. перекрывания одинаковые или противоположные знаки (рис. 1 и 3). В первом
случае взаимод. наз. связывающим, во втором-разрых-ляющим. В зависимости от
числа атомных орбиталей в сопряженной системе активир. комплекса, электронного
состояния реагирующей системы и характера вновь возникающих взаимодействий,
энергия замыкания цикла м. б. как положительной, так и отрицательной, причем
ее знак определяется без вычислений. В частности, при циклизации производных
бутадиена (р-ция 2) в основном электронном состоянии переходное состояние стабилизируется
замыканием связей посредством разрыхляющих взаимод. (перекрывание в противофазе,
рис. 3), делая энергетически выгодным конротаторный путь термич. р-ции. В первом
электронном возбужденном состоянии циклич. переходное состояние стабилизируется
связывающим взаимод. (перекрывание в фазе), что соответствует дисротаторному
пути. Эти общие положения позволяют предсказывать закономерности Р. с. согласованных
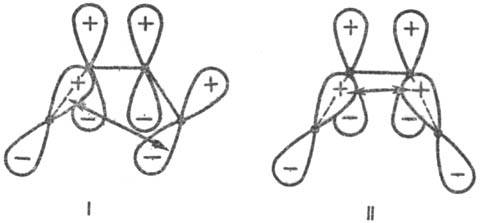
электроциклич. р-ций (Р. Вуд-ворд, Р. Хофман, 1965). Рис. 3. Переходные состояния
р-ции циклизации производных бутадиена. I-конротаторный путь р-ции (орбитали
вновь образующейся связи перекрываются в протпвофазе); II-дисротаторный путь
р-ции (орбитали перекрываются в фазе). Обозначения те же, что и на рис. 1. Стрелки
указывают области наилучшего перекрывания атомных орбиталей реакц. центра. Индексы Р. с. - теоретич. величины, к-рые используют для характеристики Р.с. на простом модельном уровне и обычно рассчитывают квантовохим. методами. Один из индексов Р. с.-энергия стабилизации; др. индексы менее универсальны, часто они являются приближенными оценками энергии стабилизации применительно к конкретным типам р-ций. Так, в случае ионных р-ций с участием p-элект-ронных систем в качестве индексов Р.с. применяют p-электронные поляризуемости (для углеводородных субстратов) и p-электронные эффективные заряды атомов (для углеводородных субстратов с полярными заместителями или для гетероциклич. субстратов). В случае радикальных р-ций присоединения в качестве индексов Р.с. особенно подходящи энергии локализации, а также индексы своб. валентности. В частности, опытные константы скорости присоединения метильного радикала к олефинам и ароматич. молекулам (рис. 2) удовлетворительно коррелируют с энергиями локализации. Совр. развитие представлений об индексах Р. с. связано с т. наз. теорий функционала плотности, согласно к-рой для квантовомех. описания электронного строения мол. систем вместо волновой ф-ции используется ф-ция электронной плотности (Р. Парр, В. Янг, 1989). Индексы Р. с. применимы
в тех случаях, когда хим. перестройка затрагивает в осн. p-электронные
системы реагентов. Если же наряду с p-электронной системой значительно
перестраиваются и s-связи реагентов, применение p-электронного приближения
мало эффективно. В подобных случаях, а также если в реагирующих системах вообще
нельзя выделить систему p-электронов, изменения энергий активации р-ций,
составляющих реакц. серию, оцениваются с помощью полуэмпирич. или неэмпирических методов
квантовохим. вычислений в рамках разл. моделей р-ции, учитывающих
все валентные электроны. В результате получают индексы Р. с., к-рые при подходящем
выборе модели удовлетворительно коррелируют с кинетич. изменениями. Такое квантовохим.
описание утрачивает качеств. наглядность p-электронной модели. Соотношение Брёнстеда .
Если простые квантовохим. методы неэффективны, Р. с. часто интерпретируют с
помощью определяемых на опыте корреляций между логарифмами констант скорости
ki и констант равновесия Кi р-ций, образующих
реакц. серию. Такие же корреляции существуют между энергиями активации Еi.
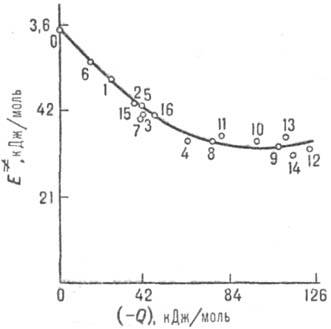
и тепловыми эффектами р-ций Qi (рис. 4). В предположении
о постоянстве пред-экспоненц. множителя в ур-нии Аррениуса для kt
оба типа корреляций эквивалентны. Типичные примеры-р-ции переноса атома
водорода или протона: (В и А-радикалы или анионы;
А···H···В-делокализован-ная структура переходного состояния) и р-ции нуклеоф.
замещения SN2:
Рис. 4. Корреляция между
энергией активации E. и тепловым эффектом Q для
газофазных р-ций отрыва атома Н метильным радикалом СН•3.
0-метан; 1-этан; 2-пропан; 3 - бутан; 4 - изобутап; 5 - неопентан; 6 - циклопропан;
7-циклогексан; 8-пропилен; 9-изобутен; 10-транс-
2-бу-тей; 11 - толуол;
12 - о-ксилол; 13- м-
ксилол; 14-n-ксилол; 15-метанол; 16-ацетон. Корреляции такого рода
часто описывают уравнением Маркуса: где lА
и lB-параметры, наз. "внутренними барьерами" реагентов
А и В. Ур-ние (6) предполагает нелинейную (квадратичную) зависимость между Е.
и Q. Однако во мн. случаях (особенно для ионно-молекулярных реакций
в полярном р-рителе) корреляции становятся прямолинейными (рис. 5), т.е.
маркусова кривизна брёнстедовских графиков исчезает.
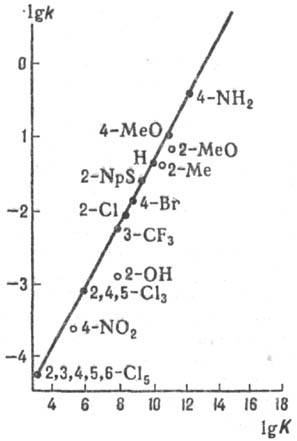
Рис. 5. Корреляция между
константами скорости k и константами равновесия К для р-ции SN2
(5) в диметилсульфоксиде при 25 °С. АСН2R - бутилхлорид (А =
С1), В- - замещенные фенилтиолят-анионы (указаны заместители), NpS
-нафтилтиолят-анион. В отличие от квантовохим.
теорий, объясняющих Р.с. исходя из электронного строения реагентов, соотношение
(6) и др. ур-ния такого типа основаны на анализе изменений структуры ППЭ в реакц.
серии. Рассматривают смещение положения переходного состояния на координате реакции
в зависимости от заместителей. Положение переходного состояния характеризуется
безразмерной величиной а, к-рая может принимать значения между нулем (положение
реагентов на координате
р-ции) и единицей (положение продуктов на координате р-ции). В рамках простейших
моделей ППЭ Производную dE.
/dQ находят из соотношений между Е. и Q типа
(6); она наз. брёнстедовским коэффициентом. Ф-ла (7) является количеств. формулировкой
постулата Хэммонда, утверждающего, что в реакц. серии переходное состояние тем
более продвинуто по координате р-ции в направлении продуктов, чем меньше экзотермич-ность
р-ции (и чем меньше Р. с.). Известны исключения, т. е. такие реакц. серии, в
к-рых неравенства (7) не соблюдаются. Они свидетельствуют о сложности строения
соответствующих ППЭ, не учтенной при выводе ф-лы (7). Влияние среды на Р.с. Совр.
развитие теории Р. с. связано с изучением влияния среды на р-ции, протекающие
в конденсир. фазе. (См. также Реакции в растворах
). Согласно классич.
подходу (С. Глесстон, К. Лейдлер, Г. Эйринг, 1941), определяют изменения энергии
начального состояния реагентов и переходного состояния под влиянием среды (р-рителя)
в рамках теории активир. комплекса. Роль среды сводится к созданию дополнит.
потенциала, модифицирующего ППЭ соответствующей р-ции в газовой фазе. Этот потенциал
м.б. учтен в совр. квантовохим. расчете. Модификация ППЭ потенц. полем среды
м. б. очень велика для р-ций с переносом заряда в полярном р-рителе, напр. SN2. С принципиальной
точки зрения наиб. интересны такие хим. р-ции, к-рые сопровождаются перестройкой
среды. Их изучение требует от теории явного рассмотрения координат, связанных
с изменениями, происходящими в среде. Иногда координата перестройки среды дает
осн. вклад в координату р-ции на энергетич. пов-сти вблизи седловой точки, соответствующей
переходному состоянию. Это означает, что потенц. барьер преодолевается в осн.
за счет движения частиц среды. Типичная модельная р-ция такого типа-внешне-сферный
перенос электрона окружении ионов (см. Одноэлектронный перенос
). Перенос
электрона фиксируется изменением состояния среды (переход из состояния I в состояние
II). Предполагается, что перенос
электрона происходит при фиксир. расстоянии между ионами и не учитываются (в
простейшем случае) структурные изменения в лигандном Для р-ций в полярном р-рителе
состояние среды характеризуется ее поляризацией, связанной со средней величиной
и ориентацией дипольных моментов молекул р-рителя. Поляризация среды (дипольный
момент, отнесенный к единице объема), если среда равновесна, определяется распределением
электрич. зарядов в хим. подсистеме и изменяется вместе с ним. Для внешнесферного
переноса электрона координата перестройки среды есть коллективная переменная-величина,
выражающаяся через поляризацию среды. В более сложных (и химически более интересных)
ситуациях координаты перестройки среды и внутр. координаты хим. подсистемы рассматриваются
на равных основаниях, т.е. энергетич. пов-сть строится в пространстве координат
обоих типов. При этом возможны неравновесные эффекты: если движение системы
по координате перестройки среды более медленное, чем движение по "химической"
координате, среда не успевает подстроиться под "химическое" движение.
Тогда имеет место неравновесная сольватация переходного состояния и теория активир.
комплекса не применима, поскольку она основана на предположении о равновесном
распределении энергии в переходном состоянии. Р. с. в таких системах коррелирует
с динамич. характеристиками среды. Для р-ций с перераспределением заряда в полярном
р-рителе динамич. характеристикой среды является время тL
продольной электрич. релаксации (рис. 6). Если же электрич. взаимодействие хим.
подсистемы со средой не столь существенно, на первый план выступают эффекты,
зависящие от гидродинамич. вязкости h среды или от коэф. диффузии D (рис. 7), обратно пропорционального вязкости (h ~ D-1).
Корреляции Р. с. с динамич. характеристиками среды находят объяснение в рамках
стохастич. кинетич. теорий.
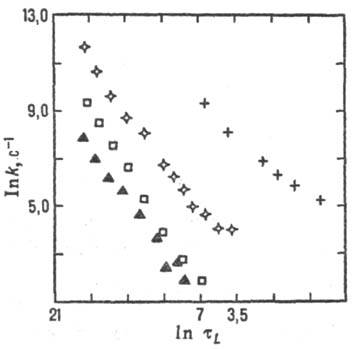
Рис. 6. Корреляция между
константами скорости k и временами продольной диэлект-рич. релаксации
тL (р-ция переноса электрона между комплексами рутения
и метилвио-летом в глицерине; значки разл. формы относятся к разл. лигандам
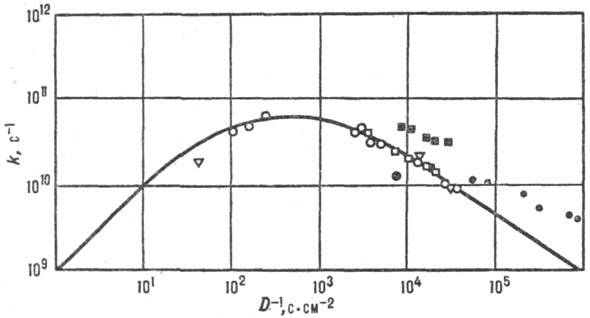
в комплексе рутения). Рис. 7. Корреляция между
константами скорости k и козф. диффузии D (фото-хим. изомеризации
стильбена в газовой и жидкой фазах при разных давлениях). Значки разл. формы
отвечают разным средам; сплошная линия—расчет по ур-нию Крамерса. Простейшая стохастич. теория,
установившая зависимость константы скорости р-ции от вязкости, была разработана
Г. Крамерсом (1940). В рамках стохастич. описания энергетика р-ции считается
известной и исследование кинетики, если р-ция равновесна, переносится на предэкспоненц.
множитель А. Если же р-ция протекает неравновесно, аррениу-совская зависимость
константьд скорости р-ции теряет смысл, т. к. энергия активации Е.
и предэкспоненц. множитель зависят от т-ры. Между "эффективными"
значениями Е и А часто наблюдаются корреляции (см. Компенсационный эффект
). Изложенные общие принципы
описания влияния среды на Р. с. перспективны для применения к хим. р-циям, протекающим
в сложных системах, напр. биологических. Разобранные примеры свидетельствуют
о многообразии проблем, связанных с Р. с. Для простейших реакц. серий достаточны
элементарные квантовохим. рассуждения (р-ции 1 и 2). В более сложных случаях
для понимания закономерностей Р. с. необходимо привлекать динамич. подходы (см. Динамика элементарного акта
) и методы статистич. физики (пример
8 и рис. 6). Вся совокупность этих проблем и применяемых при их решении методов
составляет содержание совр. теории хим. р-ций. Лит.: Базилевский М. В., Метод молекулярных орбит и реакционная способность органических молекул, М., 1969; Вудворд Р., Хоффман Р., Сохранение орбитальной симметрии, пер. с англ., М., 1971; Гаммет Л., Основы физической органической химии, пер. с англ., М., 1972; Дыоар М. Дж., Догерти Р., Теория возмущений молекулярных орбиталей в органической химии, пер. с англ., М., 1977; Салем Л., Электроны в химических реакциях, пер. с англ., М., 1985; Базилевский М. В., Колдобский С. Г., Тихомиров В. А., "Успехи химии", 1986, т. 55, в. 10, с. 1667-98; The chemical physics of solvation, pt A, ed. by R. R. Dogonadze [a. o.], pt C, ed. by A. A. Kornyshev and I. Ulstrup, Amst, 1985 88. М. В. Базилевский.
|
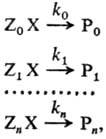
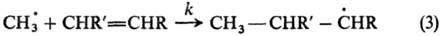
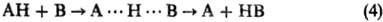
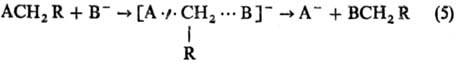
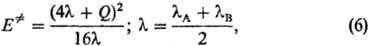
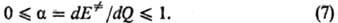

Поделитесь с друзьями:
Вы можете поставить ссылку на это слово:
будет выглядеть так: РЕАКЦИОННАЯ СПОСОБНОСТЬ
будет выглядеть так: Что такое РЕАКЦИОННАЯ СПОСОБНОСТЬ